Article Text

Abstract
Background Development of diffuse-type gastric cancer (DGC) starts with intramucosal lesions that are primarily composed of differentiated, non-proliferative signet ring cells (SRCs). These indolent lesions can advance into highly proliferative and metastatic tumours, which requires suppression of DGC cell differentiation.
Objective Our goal was to identify molecular changes contributing to the progression of indolent to aggressive DGC lesions.
Design We conducted spatial transcriptomic analysis of patient tumours at different stages of hereditary DGC, comparing transcriptional differences in tumour cell populations and tumour-associated cells. We performed functional analysis of identified changes in a human gastric (CDH1KO) organoid model recapitulating DGC initiation.
Results Our analysis reveals that distinct DGC cell populations exhibit varying levels of Wnt-signalling activity, and high levels of Wnt signalling prevent differentiation into SRCs. We identify multiple adaptations during DGC progression that converge on Wnt signalling, allowing tumour cells to remain in an undifferentiated state as they disseminate away from the gastric stem cell niche. First, DGC cells establish a cell-autonomous source for Wnt-pathway activation through upregulated expression of Wnt-ligands and ‘secreted frizzled-related protein 2’ (SFRP2) that potentiates ligand-induced Wnt signalling. Second, early tumour development is marked by extracellular matrix remodelling, including increased deposition of collagen I whose interactions with DGC cells suppress their differentiation in the absence of exogenous Wnt ligands.
Conclusions Our findings demonstrate that tumour cell-derived ligand expression and extracellular matrix remodelling sustain Wnt signalling during DGC progression. These complementary mechanisms promote niche independence enabling expansion of undifferentiated DGC cells needed for the development of advanced tumours.
- GASTRIC CANCER
- CELL DIFFERENTIATION
- EPITHELIAL CELL ADHESION
- FAMILY CANCER
- ORGANOIDS
Data availability statement
Data are available on reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information. Normalised and averaged data of the Nanostring DSP analysis has been added as a supplementary table. Raw counts of the Nanostring DSP analysis and whole exome sequencing data are available on request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
Genetic mutations underlying the initiation of diffuse-type gastric cancer (DGC) have been identified.
Patients with hereditary DGC (HDGC) often develop multiple early-stage lesions, although only a subset progresses to advanced, life-threatening tumours.
While molecular characteristics of advanced DGC have been studied, the stepwise progression of the disease remains poorly understood.
WHAT THIS STUDY ADDS
Spatial profiling of different DGC cell populations and stages reveals that Wnt signalling is crucial for the expansion of undifferentiated cells underlying HDGC progression.
Persistence of Wnt signalling is achieved independently of mutations in this pathway.
Niche independence is established through tumour cell-autonomous ligand production and remodelling of the stromal matrix that primes tumour cells for ligand-dependent Wnt-pathway activation.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
Identification of molecular mechanisms underlying stepwise DGC progression aids in understanding and predicting tumour malignancy.
Identified progression markers can help prioritise prophylactic gastrectomy for patients at high risk of developing aggressive disease and support endoscopic surveillance strategies for low-risk patients.
Insights into the biology of DGC progression may foster the development of targeted therapies and novel treatment approaches.
Introduction
Gastric cancer is the fifth-leading cause of cancer-related deaths worldwide, with diffuse-type gastric cancer (DGC) accounting for a third of diagnoses.1 2 For individuals with pathogenic germline variants of CDH1 or CTNNA1 at risk for developing hereditary DGC (HDGC), prophylactic gastrectomy at an early age is currently recommended.3 Resected stomachs from HDGC patients typically show multiple asymptomatic lesions confined to the gastric mucosa. However, recent epidemiological studies suggest that only 10%–40% of HDGC patients develop life-threatening advanced tumours.4 5 Therefore, gastrectomy would preferably be reserved for patients with potentially aggressive DGC. Before such a paradigm shift can be implemented, it is crucial to deepen our understanding of the mechanisms driving early lesion progression to advanced tumours and to improve our ability to predict tumour malignancy.
Early-stage lesions originate from cells escaping from the proliferative isthmus region of gastric glands into the stroma.6 7 These indolent lesions predominantly consist of differentiated, mucus-filled signet ring cells (SRCs) and a small population of proliferative, poorly differentiated cells (PDCs) at the lesion base.6 8 Progression into advanced DGC requires expansion of this PDC population and these cells eventually infiltrating beyond the mucosa (figure 1A).6 9–11 Cells that escaped from the gastric epithelium may initially retain access to stem cell niche factors present in the isthmus region, explaining why cells at the base of initial lesions can reside in an undifferentiated state.11 However, as cells disseminate further into the mucosa and lose access to these niche factors, they will need to overcome the dependency on the gastric stem cell niche to sustain an undifferentiated and proliferative state.6 10 11

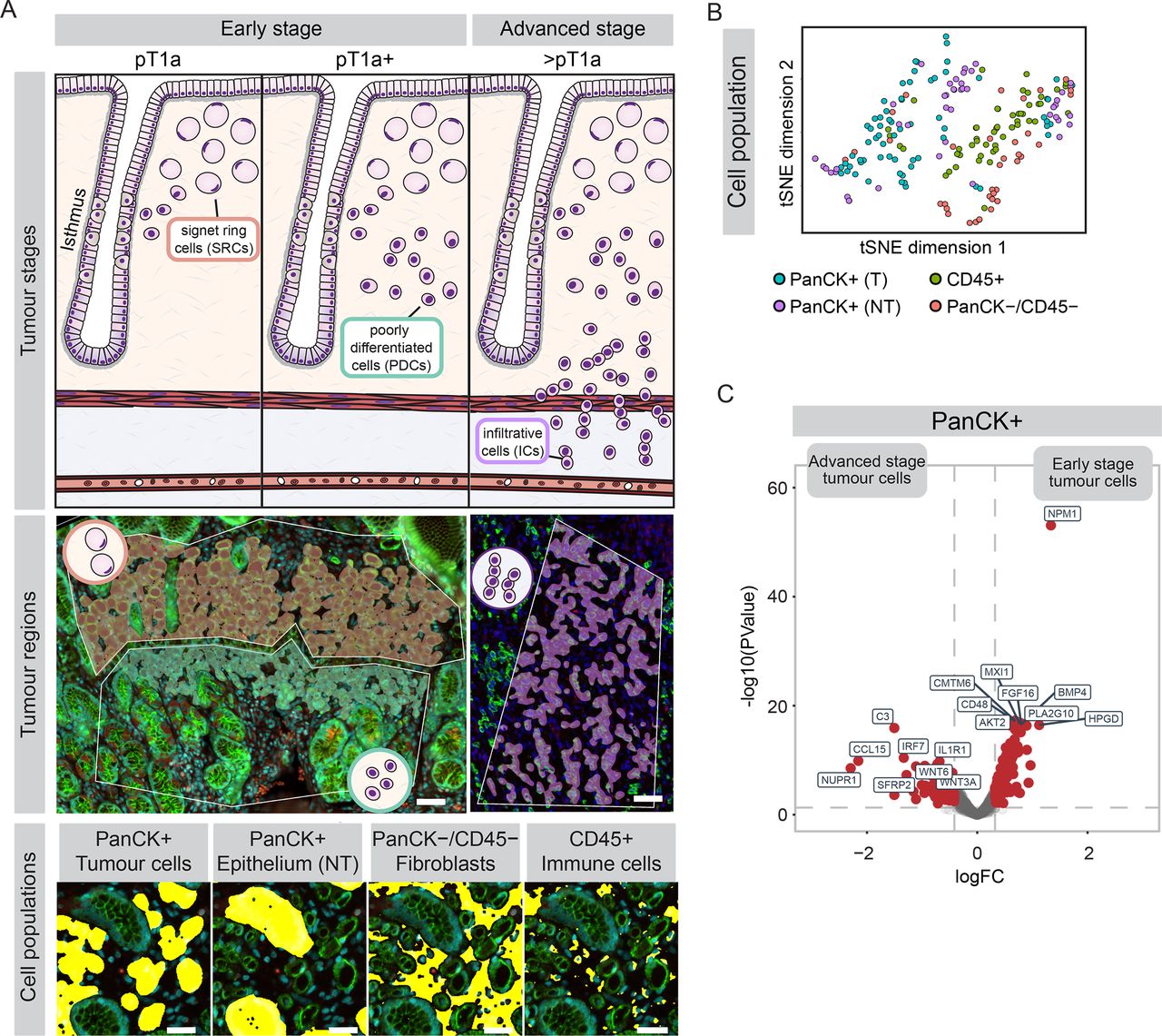
Spatial transcriptomic analyses of DGC cell populations across tumour stages. (A) Top: schematic representation of early-stage (pT1a or pT1a+) and advanced-stage (>pT1a) DGC. Middle: overlays of tumour regions containing signet ring cells (SRCs, orange), poorly differentiated cells (PDCs, teal) and infiltrative cells (ICs, purple), projected on tissue stainings (for PanCK, green; CD45, red and Cyto-13, cyan) of pT1a lesion (left) and pT3 lesion (right). Scalebar: 50 µm. Bottom: segmentation of cell populations (yellow) within tumour regions, containing tumour cells (CD45−/PanCK+), surrounding non-tumourous (NT) gastric epithelium (CD45−/PanCK+; separated based on morphology), immune cells (CD45+/PanCK−) and non-immune stromal cells (predominantly fibroblasts; CD45−/PanCK−). Scalebar: 30 µm. (B) t-SNE of integrated transcriptomic datasets of different DGC cell populations, coloured by cell type. (n=200 regions). (C) Differential gene expression in PanCK+ tumour cells comparing early-stage (pT1a and pT1a+; n=21 regions from 7 patients) to advanced-stage (>pT1a; n=44 regions from 7 patients) lesions. DGC, diffuse-type gastric cancer; PanCK, Pan-cytokeratin; T, tumourous.
Loss of functional E-cadherin is a primary initiating event in both sporadic and HDGC.3 12 E-cadherin loss induces the formation of SRC lesions in mice, yet is insufficient for progression to advanced DGC.13 Additional genetic alterations, including mutations in TP53 or RHOA that promote tumour cell survival, are linked to advanced DGC development.12 14 15 Gastric organoid models demonstrated that mutations in growth factor pathways, including Wnt and MAPK pathways, can establish niche independency of DGC cells.16 However, mutations in components of these niche factor signalling pathways are rare in human DGC,12 implying that other mechanisms contribute to niche independency.
Previous studies investigating molecular changes associated with DGC development have primarily focused on the analysis of advanced DGC tumours,17–21 and the stepwise progression of DGC remains poorly understood. Here, we performed spatial transcriptional analysis of distinct cell populations and stages of HDGC. In combination with functional characterisation of these changes in DGC organoid models, we uncovered how niche independence is established during DGC progression.
Materials and methods
Nanostring Geomx digital spatial profiling
Tumour microarrays (TMAs) were constructed using formalin-fixed paraffin-embedded tissue samples from 38 DGC lesions from 13 patients harbouring a confirmed germline pathogenic variant in the CDH1 gene. These samples were collected from Radboud university medical centre (Radboudumc), Antoni van Leeuwenhoek Hospital and UMC Utrecht (online supplemental table 1). A waiver of consent was granted for this study, ensuring that data from patients who had opted out of research use were excluded. GeoMx digital spatial profiling (DSP) was performed on TMAs according to the manufacturer’s instructions and as previously described.22 Additional information on DSP labelling and analysis, as well as whole exome sequencing of advanced tumours from this tumour panel, can be found in online supplemental methods. Averaged normalised counts of GeoMx DSP can be found in online supplemental table 2).
Supplemental material
Supplemental material
Supplemental material
Organoid culture
Human gastric CDH1KO organoids7 were cultured at 37°C and 5% CO2 in basement membrane extract (BME; 3533-010-02; Bio-Techne) in advanced DMEM/F12 medium (Invitrogen) containing gastric organoid medium (composition can be found in online supplemental methods). Organoids were passaged as single cells using trypsin–EDTA (T3924; Sigma-Aldrich) once a week and grown for the first 2days in the presence of Y-27632 (10 µM; M1817). Organoids were kept in culture for a maximum of 15 passages.
For single cell seeding in BME and collagen I, organoids were isolated from BME cultures using 1 mg/mL dispase II (Cat# 17105041, Life Technologies), followed by three washes with DMEM/F12 media and dissociation in phosphate buffer saline (PBS) containing 2 µM EDTA. Cells were then either embedded in BME or in type I collagen gels consisting of non-pepsinised rat-tail Collagen I (Corning, Cat# 354236) at a final concentration of 1 mg/mL, in gastric organoid medium lacking Y-27632. The collagen I mixture was prepared in buffer containing 10X PBS (Gibco), NaOH and dH2O, and pre-polymerised on ice for 2–3 hours. Embedding of cells in BME and collagen I gels was followed by incubation at RT for 5 min, plating as 10 µL drops in eight-well imaging dish (µ-Slide, Ibidi) or six-well plates and incubation at 37 C° until full polymerisation of the gels was achieved. For experiments that include the use of inhibitors, DMSO, BTT-3033, LGK974, F2.I or iCRT3 were added immediately after plating. For functional analyses of organoids by immunostainings, flow cytometry, live imaging and RT-qPCR see online supplemental methods.
Patient involvement statement
Patient representatives from the Dutch HDGC foundation ‘Stichting CDH1’ contributed to reviewing the methods and associated funding application. Patient priorities were explored during biannual HDGC patient days, where the authors (RSvdP, TMB, JMvD and LLK) engaged with patients through workshops and presentations. Most HDGC patients in the Netherlands are treated by the two gastroenterologists (TMB and JMvD) authoring this manuscript, integrating their clinical insights into the research. Study findings were shared with patients at national and international workshops. Patients strongly support this research, seeking safe alternatives to prophylactic gastrectomy and deeper insights into (H)DGC’s molecular mechanisms.
For additional information, see online supplemental methods.
Results
Spatial transcriptomic analyses of DGC cell populations across tumour stages
To investigate molecular changes underlying the progression from indolent early-stage DGC towards infiltrative tumours, we conducted spatial transcriptomic analysis of different stages of HDGC tumours. We included 12 early-stage (7 pT1a, 5 pT1a+; early lesions with increased proportion of PDCs) and 7 advanced-stage (>pT1a) tumours (figure 1A),9 from 13 HDGC patients with confirmed germline pathogenic variants of CDH1. Using H&E staining, tumours were subdivided into regions containing the distinct cell populations; SRCs, PDCs and infiltrative cells (ICs) that infiltrated beyond the mucosa (figure 1A). Tumour slides were stained with nuclear (SYTO13), epithelial (PanCK) and immune cell (CD45) markers, allowing for further discrimination between tumour cells, non-tumourous gastric epithelium, immune cells and non-immune cells present in the stroma (predominantly fibroblasts23) (figure 1A). Furthermore, we included regions containing gastric glands distant from tumour lesions, to enable comparison of stromal cells in non-tumourous areas with those in tumour-associated stroma. Subsequently, we profiled RNA expression in cell populations across individual tumour regions using the Nanostring GeoMx platform.24 Unbiased t-SNE analysis showed that PanCK-positive cells cluster separately from immune cells and non-immune stromal cells (figure 1B). Furthermore, markers specific to epithelial cells, fibroblasts and immune cells were selectively expressed within their respective cell clusters (online supplemental figure 1A-C). We did not observe any clustering based on patients and/or tissue slides, excluding a contribution of potential confounding factors on further analyses (online supplemental figure 1D, E).
Supplemental material
We next compared the expression profiles of the entire tumour cell population across different DGC stages, which revealed minimal gene expression differences between pT1a and pT1a+ tumours (online supplemental figure 1F, G). However, comparing early-stage lesions (pT1a and pT1a+) with advanced-stage DGC showed prominent transcriptional differences (figure 1C, online supplemental table 4). This included multiple transcriptional changes in advanced DGC that were previously reported to mark DGC progression, for example, upregulation of NUPR1 and IRF7 and downregulation of NPM1 (figure 1C).25–27 Thus, our transcriptional profiling indicates that the most prominent changes in gene expression in DGC cells occur during the transition into advanced-stage tumours.
Supplemental material
Wnt signalling sustains the undifferentiated state of tumour cells throughout DGC progression
As DGC progression relies on the expansion of undifferentiated cells, we compared the transcriptional profile of differentiated SRC cells with undifferentiated PDC and IC populations in advanced DGC (figure 2A, B, online supplemental table 5 and 6). Approximately 30 genes were significantly upregulated in PDCs and over 50 genes in ICs compared with SRCs. Upregulated genes in both PDCs and ICs include genes previously reported to correlate with poor prognosis (eg, MMP7, CD24, DMBT1).25–27
Supplemental material
Supplemental material

Wnt signalling maintains tumours cells in an undifferentiated state throughout DGC progression. (A, B) Differential gene expression in PanCK+ tumour cells comparing SRCs (n=7 regions from 4 patients) to PDCs (n=10 regions from 6 patients) (A) or to ICs (n=27 regions from 7 patients) (B) of advanced-stage DGC lesions. (C) Comparison of Wnt-signalling signatures in SRCs to PDCs (top) and ICs (bottom). P-adj. is represented by colour code, number of genes in geneset by dot size. See online supplemental figure 2A) for expression of individual Wnt targets from these signatures. (D) Representative phase-contrast images with DAPI staining of CDH1KO organoids, 5 days after seeding as single cells in the presence or absence of Wnt-surrogate (Wnt) and R-spondin 1 (RSPO1) in BME. Scalebar: 30 µm. (E) Representative images of H&E, KI67 and MUC5AC staining on serial sections of pT1a stage (top) and pT1a+ stage (bottom) DGC lesion. Examples of SRCs and PDCs are highlighted by orange and teal arrowheads, respectively. Scalebar: 50 µm. (F) Schematic representation of analysis of Wnt-dependent cell differentiation of CDH1KO gastric cells. (G) Representative images of CDH1KO cells 5 days after seeding as single cells in the presence or absence of Wnt/RSPO1 in BME and immunostained for MUC5AC with DAPI and phalloidin. Scalebar: 20 µm. (H, I) Quantification of percentage MUC5AC-high cells 5 days after seeding in presence or absence of Wnt/RSPO1 in BME, determined by microscopic analysis of single cells (H) or flow cytometry (I). **p<0.005 (J) Quantification of percentage of KI67-high cells 5 days after seeding in presence or absence of Wnt/RSPO1 in BME, determined by flow cytometry. **p<0.005. BME, basement membrane extract; DGC, diffuse-type gastric cancer; IC, infiltrative cell; PanCK, Pan-cytokeratin; PDCs, poorly differentiated cells; SRCs, signet ring cells.
Supplemental material
To uncover signalling cascades that could promote DGC cells to reside in an undifferentiated state, we performed unbiased gene set enrichment analysis (figure 2C).28 This revealed a significant enrichment of the ‘TCF-dependent Wnt signalling’ gene signature in ICs compared with SRCs (figure 2C, online supplemental table 7). More specific analysis, by comparison to published ‘APC-KO’ signatures’ derived from cells in which the Wnt pathway is constitutively active, verified upregulation of Wnt-target genes both in the PDC and IC populations (figure 2C and online supplemental figure 2A).29 30 Thus, our data imply that PDCs and ICs exhibit higher activity of the Wnt-signalling pathway, which is known to promote self-renewal and proliferation of gastric epithelial stem cells and DGC cells.16 31 32
Supplemental material
To further investigate whether Wnt signalling controls the fate of cells that escaped from the gastric epithelium, we used a CDH1-depleted human gastric organoid model (CDH1KO) that recapitulates the initiation of DGC.7 These organoids are typically cultured in the presence of Wnt-surrogate (Wnt) and R-spondin 1 (RSPO1), stimulating activation of the Wnt receptor through heterodimerisation with its coreceptor and controlling its plasma membrane abundance, respectively.33–35 When seeding single cells derived from CDH1KO organoids into a BME matrix in medium containing Wnt/RSPO1, individual cells were able to develop into multicellular organoids within 5 days (figure 2D). In contrast, in cultures deprived of Wnt/RSPO1, the single-cell organisation was retained, and cells typically displayed an SRC-like morphology with large cytoplasmic vacuole and crescent-shaped nucleus (figure 2D). In patient tumours, SRCs are typically characterised by absence of KI67 expression and accumulation of mucins, including MUC5AC, in contrast to PDCs and ICs that are mostly KI67-positive and display low mucin expression6 (figure 2E). Microscopic and flow cytometry-based analysis showed that ~50% of CDH1KO cells similarly displayed high levels of MUC5AC expression and loss of KI67 expression following 5 days in Wnt/RSPO1 deprived medium (figure 2F–J, online supplementalfigure 2B-E). This differentiation into MUC5AChigh/KI67low cells was also observed after direct inhibition of the Wnt receptor Frizzled (online supplemental figure 2F,G). These findings demonstrate that Wnt signalling governs the differentiation status of DGC cells, with cells differentiating into non-proliferative, SRC-like cells in the absence of externally provided Wnt ligands.
sFRP2-potentiated Wnt3A-signalling promotes the undifferentiated state of DGC cells
While Wnt signalling is elevated in both PDCs and ICs, oncogenic mutations within the Wnt pathway are rare in DGC.12 In line with this, in advanced DGC lesions where our spatial transcriptomic analysis indicated active Wnt signalling, whole exome sequencing did not reveal somatic mutations in components of the Wnt pathway (online supplemental figure 3A). This implies that DGC cells residing outside of the Wnt niche rely on alternative mechanisms to become niche independent and resist differentiation towards SRCs.
Supplemental material
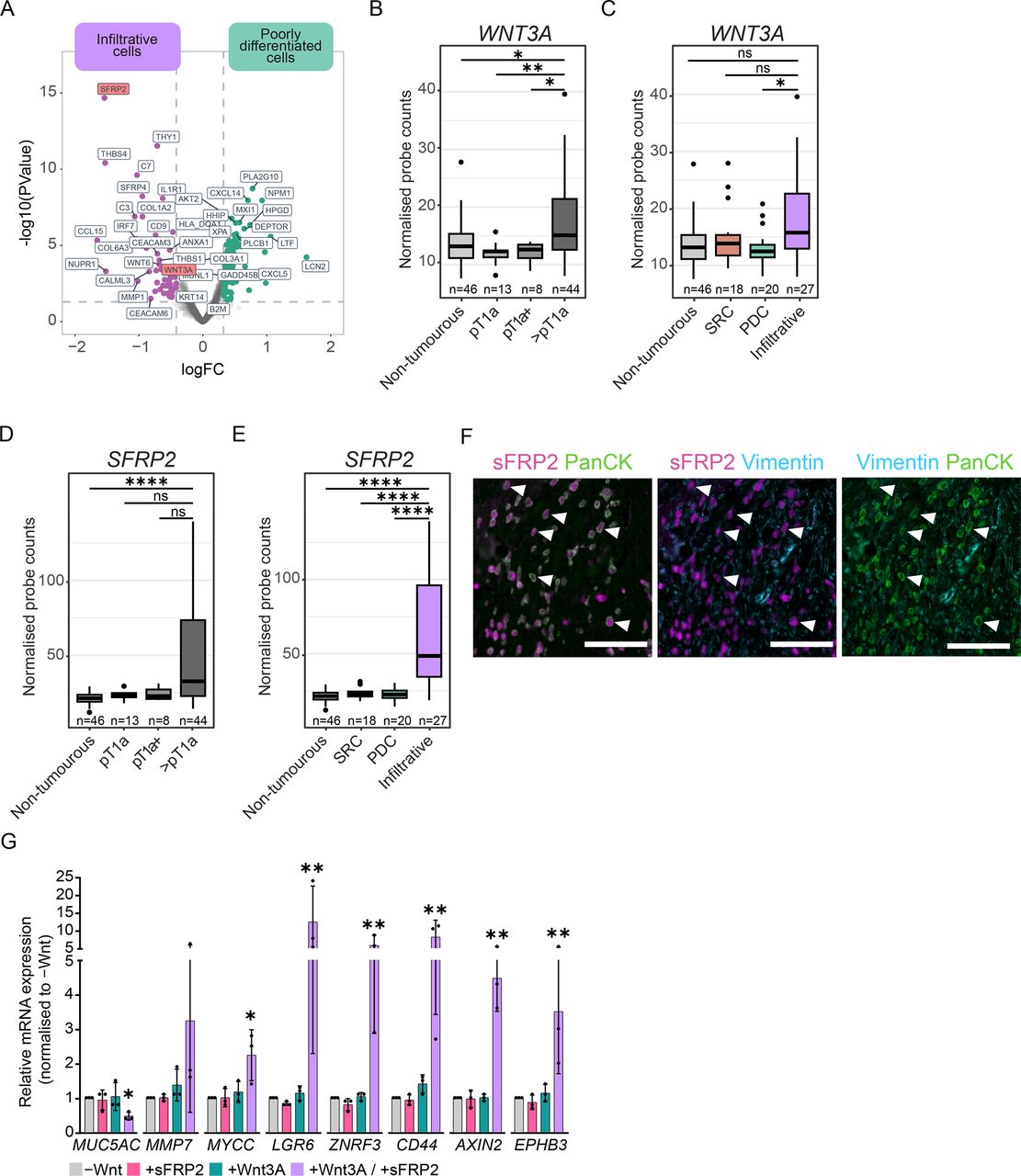
To uncover the potential underlying mechanism(s) that promote Wnt signalling in advanced DGC lesions, we examined transcriptional changes that could impact this pathway. We noted significant upregulation of the Wnt receptor ligands WNT3A and WNT6 in advanced DGC (figure 1C), specifically within the IC population and consequently increased with T-stage (figures 3A–C, online supplemental figure 3B and online supplemental table 8). Concurrently, ICs showed strong upregulation of genes encoding the Secreted Frizzled Related Proteins, particularly of SFRP2, a modulator of Wnt-ligand function (figure 1C, figures 3A, D, E, online supplemental table 8).36 Analysis of public transcriptome datasets correlated high SFRP2 expression with poor survival in DGC patients (online supplemental figure 3D).37 SFRP2 was previously shown to be upregulated in other epithelial tumours; however, in these cases, it is reported to originate from cancer-associated fibroblasts.38 To validate our spatial transcriptomic data, we performed immunofluorescent stainings, which confirmed sFRP2 expression in PanCK-positive ICs and not fibroblasts marked by Vimentin expression (figure 3F).
Supplemental material

sFRP2-potentiated Wnt3A-signalling maintains DGC cells in an undifferentiated state. (A) Differential gene expression in PanCK-positive tumour cells comparing PDCs (n=20 regions from 12 patients) to ICs (n=27 regions from 7 patients). (B, C) Normalised mRNA expression of WNT3A across tumour stages (B) and tumour cell populations (C). *p<0.05, **p<0.005. (D, E) Normalised mRNA expression of SFRP2 across tumour stages (D) and tumour cell populations (E). ****p<0.0001 (F) Representative immunostaining of infiltrative region of pT4 lesion for sFRP2, PanCK and Vimentin. Arrowheads indicate examples of sFRP2-expressing, PanCK-positive ICs. Scalebar: 100 µm. (G) RT-qPCR analysis of CDH1KO organoids cultured for 2 days in the presence of RSPO1 and either absence of exogenous Wnt, presence of recombinant Wnt3A (100 ng/mL), recombinant sFRP2 200 ng/mL or both Wnt3A and sFRP2. *p<0.05, **p<0.005. For comparison to maximum Wnt-target gene expression using Wnt-surrogate, see online supplemental figure 3F. DGC, diffuse-type gastric cancer; ICs, infiltrative cells; PDCs, poorly differentiated cells.
Previously, distinct functions of sFRPs have been described across tumour types, either facilitating Wnt3A interaction with Frizzled receptors or repressing Wnt-pathway activity.36 To explore the interplay of sFRP2 with Wnt3A in DGC cells, we plated CDH1KO organoids as single cells in the presence of recombinant Wnt3A, at concentrations sufficient to sustain organoid outgrowth but below maximum activation of the Wnt pathway (online supplemental figure 3E). Under these conditions, we tested how supplementing the medium with recombinant sFRP2 affects the activation of downstream Wnt-target genes, as well as cellular differentiation status based on MUC5AC expression. RT-qPCR analysis showed minimal upregulation of Wnt-target genes by low concentrations of Wnt3A, and no changes in their expression in the presence of only sFRP2 (figure 3G). However, combining Wnt3A and sFRP2 significantly enhanced Wnt target gene expression and resulted in reduced expression of MUC5AC, indicating that sFRP2 potentiates Wnt3A in halting DGC cell differentiation (figure 3G, online supplemental figure 3F). Together, these data demonstrate that at later stages of DGC progression, ICs upregulate expression of Wnt ligands and sFRPs, which provides a potential tumour cell-autonomous mechanism to sustain an undifferentiated state, independent of the gastric epithelial niche.
Remodelling of extracellular matrix composition during early-stage DGC progression
Although Wnt3A and sFRP2 upregulation may promote niche independence, this appears restricted to ICs that infiltrated beyond the mucosa (figure 3A, C, E). In contrast, our transcriptional analysis did not reveal apparent changes linked to Wnt signalling in tumour cells within intramucosal lesions (figures 2A and 3B–E). Therefore, we expanded our focus to examine transcriptional changes associated with DGC progression in PanCK-negative tumour-associated cells. Given the limited number of regions with sufficient CD45-positive cells for reliable analysis, we concentrated on PanCK−/CD45− cell populations consisting predominantly of fibroblasts.
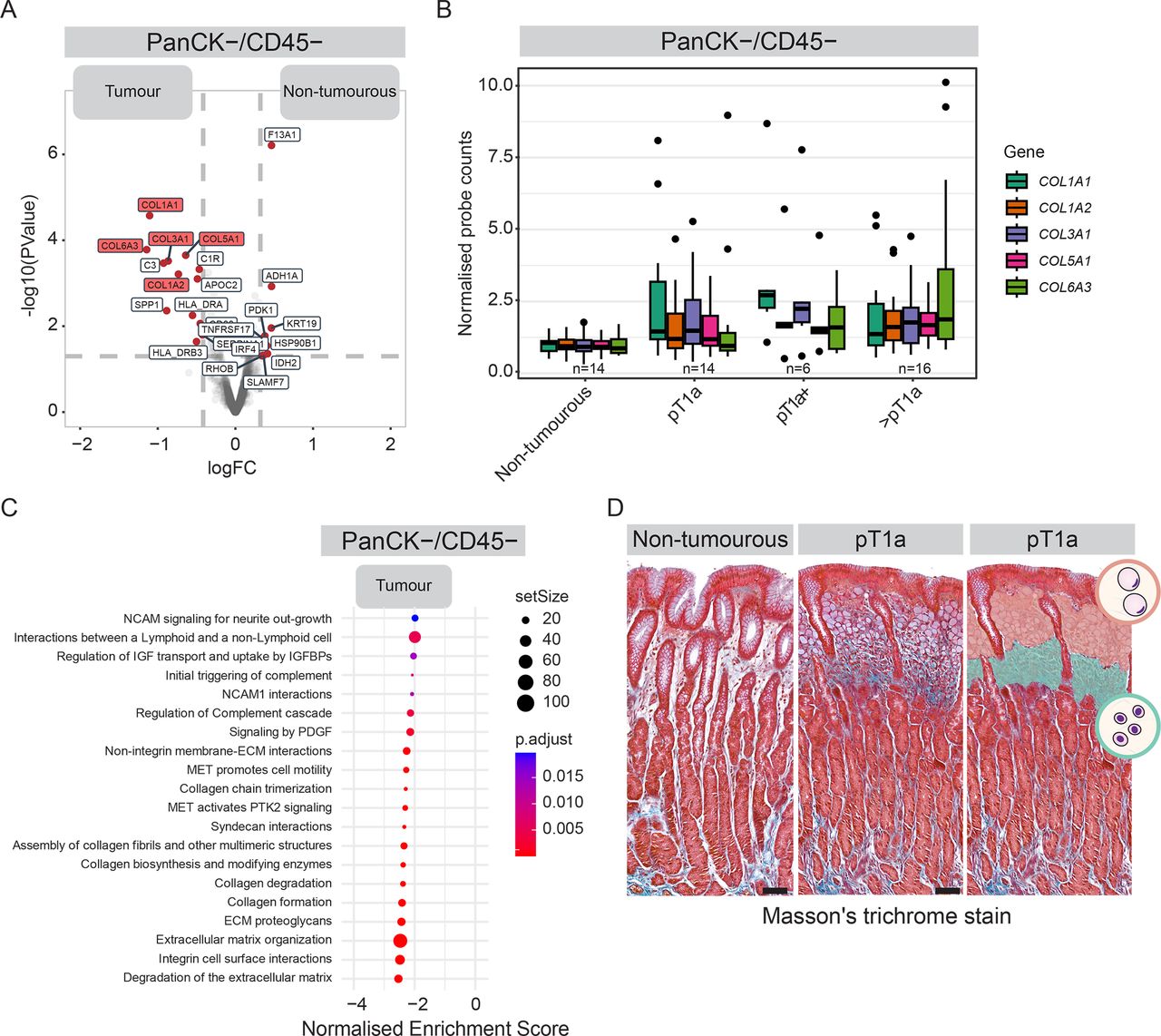
We noted strong upregulation of genes encoding for collagen proteins in PanCK−/CD45− cell populations within tumour lesions compared with non-tumourous gastric tissue (figure 4A). While expression of individual collagen genes differed between DGC tumour stages, collagen expression was consistently elevated across all stages compared with non-tumourous epithelial microenvironment (figure 4B, online supplemental figure 4A). We did not detect an increase in CD45-negative stromal cell numbers in early lesions, suggesting collagen upregulation stems from elevated expression in tumour-associated fibroblasts rather than an influx of fibroblasts (online supplemental figure 4B). In line with collagens being among the most prominently upregulated genes in CD45-negative stromal cells in DGC lesions, unbiased gene set enrichment analysis revealed extracellular matrix (ECM) remodelling to be among the top upregulated pathways in this cell population (figure 4C, online supplemental table 9).28
Supplemental material
Supplemental material

Remodelling of ECM composition during early-stage progression of DGC. (A) Differential gene expression comparing PanCK−/CD45− cells in the stroma of tumour regions (n=41 regions from 12 patients) and non-tumourous glands (n=16 regions from 10 patients). (B) Normalised mRNA expression of collagen genes in PanCK−/CD45− cells in the stroma of non-tumourous epithelium and across tumour stages. (C) Gene signatures enriched in PanCK−/CD45− cells in the stroma of tumour lesions compared with non-tumourous glands, using gene sets from the Reactome database.28 P-adj is represented by colour code, number of genes in geneset by dot size. (D) Representative Masson’s trichrome staining of the collagen network (blue) of pT1a-stage DGC lesion (middle planel) and non-tumourous region (left panel) from the same patient (p4). Right panel shows segmentation of stroma with SRC and PDC cell populations in orange and teal, respectively. Scalebar: 100 µm. See online supplemental figure 4C for examples in additional patients. ECM, extracellular matrix; DGC, diffuse-type gastric cancer; PanCK, Pan-cytokeratin; PDCs, poorly differentiated cells; SRCs, signet ring cells.
To further evaluate ECM alterations in DGC, we performed Trichrome staining to visualise collagen fibres in patient tissues. The stroma surrounding early mucosal lesions displayed increased collagen density compared with non-tumourous regions within the same patient (figure 4D). Collagen was particularly enriched around PDC cells, which was consistent across patients (figure 4D, online supplemental figure 4C). In advanced DGC, regions with PDCs showed a similar increase in collagen density compared with stroma surrounding SRCs, which persisted in regions containing ICs (online supplemental figure 4D). These findings indicate that collagen upregulation and increased deposition in the stromal matrix begin early in DGC progression and continue in advanced stages.
Collagen I interactions confer DGC cell independence from external Wnt ligands
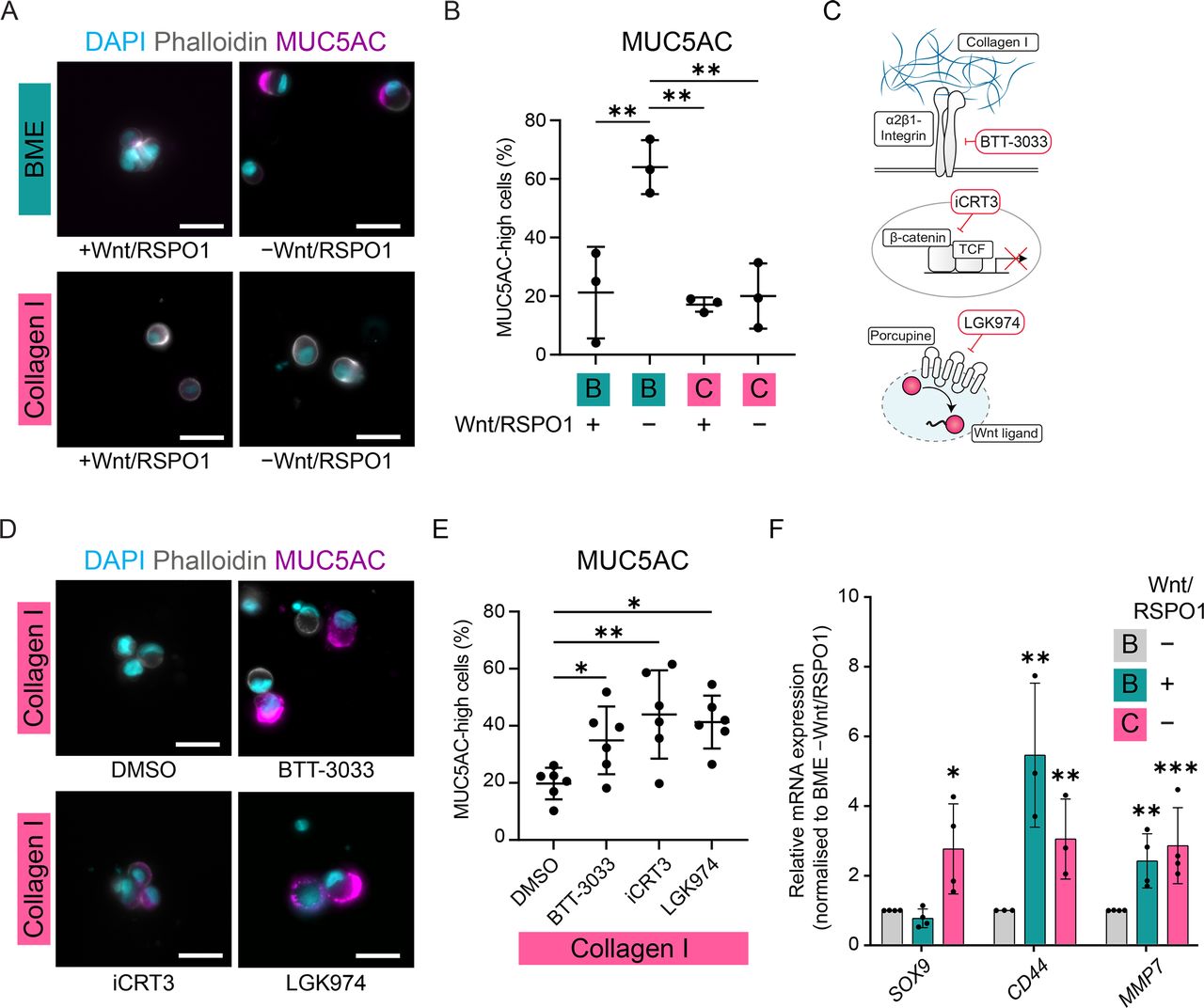
The increased abundance of collagens, particularly collagen I (COL1A), in regions with PDCs suggests this ECM remodelling may facilitate the undifferentiated state of DGC cells. In line with this, elevated expression of collagen I and other matrix genes we found upregulated in DGC correlates with poor prognosis (online supplemental figure 5A). To explore whether this change in ECM composition impacts DGC cell differentiation, we seeded single CDH1KO cells in a collagen I matrix. In the presence of Wnt/RSPO1, CDH1KO cells embedded in both collagen I and BME displayed a similar undifferentiated morphology and absence of MUC5AC expression, although cells in collagen I typically remained more dispersed (figure 5A, B). However, whereas in BME the absence of Wnt/RSPO1 resulted in differentiation into SRC-like cells expressing MUC5AC, in collagen I gels cells retained an undifferentiated state without these exogenously provided niche factors (figure 5A, B). To test whether this relies on specific interactions of DGC cells with collagen I, we functionally blocked the main collagen I-interacting integrin subtype, α2β1-integrin (figure 5C).39 BTT-3033-mediated α2β1-integrin inhibition resulted in differentiation of Collagen I-embedded cells, indicated by their SRC-like morphology and upregulation of MUC5AC (figure 5D, E). Thus, integrin-dependent interactions with collagen I enable DGC cells to sustain an undifferentiated state without exogenously supplied Wnt ligands.
Supplemental material

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Collagen I interactions confer DGC cell independence from external Wnt ligands. (A) Representative images of CDH1KO organoids 5 days after seeding as single cells in BME or Collagen I in the presence or absence of Wnt/RSPO1 and immunostained for MUC5AC with DAPI and phalloidin. Scalebar: 25 µm. (B) Quantification of percentage MUC5AC-high cells 5 days after seeding in the presence or absence of Wnt/RSPO1 in BME or collagen I, determined by microscopic analysis of single cells. **p<0.005. (C) Schematic representation of the mechanisms of action of BTT-3033 (blocking interaction of α2β1-integrin with Collagen I), iCRT3 (blocking β-catenin binding to its transcription factor TCF) and LGK974 (inhibiting Porcupine-mediated Wnt ligand palmitoylation and thereby secretion). (D) Representative images of cells of CDH1KO organoids 5 days after plating in Collagen I in absence of Wnt/RSPO1 and addition of DMSO, iCRT3, LGK974 or BTT-3033, and immunostained for MUC5AC with DAPI and phalloidin. Scalebar: 25 µm. (E) Quantification of the percentage of MUC5AC-high cells 5 days after seeding in the absence of Wnt/RSPO1 in Collagen I and addition of DMSO, iCRT3, LGK974 or BTT-3033, determined by microscopic analysis of single cells. *p<0.05, **p<0.005. (F) RT-qPCR of CDH1KO organoids cultured for 1 day in the presence or absence of RSPO1 and Wnt-surrogate (Wnt) in BME or collagen I. *p<0.05, **p<0.005, ***p<0.0005. Expression data of additional Wnt-target genes can be found in online supplemental figure 5B. BME, basement membrane extract; DGC, diffuse-type gastric cancer.
The lack of reliance on external Wnt ligands could be attributed either to collagen I interactions preventing DGC cell differentiation through a Wnt-independent mechanism, or inducing Wnt signalling in the absence of exogenous ligands. RT-qPCR analysis showed that a subset of Wnt-target genes (MMP7, CD44 and SOX9) were significantly upregulated in cells cultured in collagen I compared with BME, in absence of exogenous Wnt ligands (figure 5F, online supplemental figure 5B). Notably, these targets were among the genes we found upregulated in PDCs and ICs in our spatial transcriptomic analysis (figure 2A, B). Since these findings support the hypothesis that Collagen I interactions impinge on the Wnt pathway, we directly tested whether DGC cells interacting with collagen I continue to rely on Wnt-signalling to sustain their undifferentiated state. To this end, we inhibited Wnt-driven transcription by blocking the interaction between the Wnt pathway effector β-catenin and its transcription factor TCF using iCRT3 (figure 5C).40 Collagen I-interacting cells failed to maintain an undifferentiated state in the presence of iCRT3, resulting in the emergence of MUC5AChigh cells with SRC-like morphology (figure 5D, E). Thus, while cells sustain their undifferentiated state within a collagen I matrix without exogenous Wnt ligands, they remain dependent on Wnt-driven transcriptional activity.
Because Wnt ligands are essential for activating Wnt signalling, potentially, cells in collagen I could provide their own Wnt ligands. To test this, we blocked ligand synthesis by targeting the Porcupine enzyme, which mediates Wnt ligand palmitoylation that is indispensable for its secretion (figure 5C).41 Similar to inhibition of Wnt-mediated transcription, the presence of the Porcupine inhibitor LGK974 resulted in the differentiation of DGC cells cultured in collagen I in absence of Wnt/RSPO1 (figure 5D. E). These data demonstrate that collagen I interactions confer DGC cell independence from external Wnt ligands to sustain an undifferentiated state, which is dependent on the production of endogenous Wnt ligands. Altogether, our findings demonstrate that Wnt signalling dictates the differentiation status of DGC cells and identified both tumour cell-intrinsic and tumour-microenvironmental changes during DGC progression that promote Wnt signalling and could establish niche independence to resist differentiation towards non-malignant SRCs.
Discussion
DGC progression involves the infiltration of tumour cells beyond the mucosa, which requires expansion of the population of proliferative PDCs by preventing their differentiation into SRCs. Using spatial transcriptomic profiling of HDGC lesions and a CDH1KO gastric organoid model, we demonstrate that Wnt signalling activity is a key determinant of DGC cell fate and essential to sustain an undifferentiated cellular state during tumour progression. ICs that disseminated beyond the mucosa themselves produce Wnt ligands, enabling autocrine activation of the Wnt pathway that is potentiated by sFRP2 expressed by these cells. Starting in the early stages of DGC, the stromal matrix is remodelled, including increased deposition of collagen I whose interaction with DGC cells suppresses their differentiation into SRCs in the absence of exogenously provided Wnt ligands. Together, these complementary mechanisms that converge on Wnt signalling allow disseminating cells to resist differentiation while being spatially segregated from the gastric epithelial stem cell niche. Although our analysis focused on HDGC, the histological similarity between sporadic and HDGC tumours suggests that our findings are similarly applicable to sporadic DGC.11
Despite the importance of niche independence, mutations in the Wnt pathway, common in carcinomas, are relatively rare in DGC.12 Indeed, in advanced DGC lesions with persistent Wnt signalling activity (figure 2C), we did not detect any mutations in Wnt-pathway components (online supplemental figure 3A). This observation may align with the ‘just-right’ model, which suggests that submaximal levels of Wnt activation are sufficient to drive transformation, whereas excessive activation can be cytotoxic and reduce tumour-promoting effects.42 Moreover, the level of Wnt signalling may influence the histological growth pattern of gastric cancer, as genetic mutations in the Wnt pathway are more frequently associated with tubular growth pattern in both humans and mice models.43–45 Although Wnt pathway mutations are rare in DGC, genetic alterations are still shown to influence niche independence. Phenotypic analysis of patient-derived and genetically engineered organoids revealed that compound mutations in CDH1 and TP53 result in independence from exogenously provided R-spondin to activate the Wnt pathway.16 While these tumour cells do not require R-spondin, which regulates internalisation and degradation of Wnt receptors, they remain dependent on external Wnt ligands for activation of this pathway.34 35
Our findings reveal that DGC lesion expansion beyond the mucosa is closely associated with the upregulation of Wnt ligands and sFRP2. This suggests a tumour cell-autonomous mechanism for ligand-dependent activation of the Wnt pathway, which is potentiated by sFRP2 to maintain pathway activity even in conditions of lower ligand concentrations. This potentiation of Wnt signalling by sFRP2 observed in DGC cells may involve stabilisation of Wnt ligands or an increase in their binding affinity for receptors.38 46–48 Our identification of the production of Wnt ligands and sFRP2 by DGC cells complements earlier studies describing other sources of ligands activating the Wnt pathway, including stromal fibroblasts at the gland base producing R-spondin32 49 and mucosal innate lymphoid cells producing Wnt5A on chronic inflammation in mice.31 50 Importantly, our study emphasises that DGC cells themselves are capable of producing Wnt ligands, allowing them to regulate Wnt signalling autonomously, even as they invade beyond the mucosa.
Our identification of a role for ECM remodelling in DGC progression advances on previous studies showing accumulation of ECM components in advanced lesions compared with earlier DGC stages and healthy tissue in patients and mouse models.51–55 Our transcriptomic and histological analyses demonstrate that ECM remodelling begins as tumour cells invade the stromal compartment, with increased expression of collagen I and other matrix proteins in stromal fibroblasts and enhanced collagen deposition, particularly in the stroma surrounding PDCs and ICs. Our gastric organoid model shows that collagen I interactions diminish the dependence on exogenous Wnt ligands to activate the Wnt pathway and block differentiation. Importantly, cells remain reliant on Wnt signalling, as inhibiting secretion of Wnt ligands by tumour cells leads to their differentiation. Integrin signalling may enhance the sensitivity of DGC cells to Wnt ligands, which could be produced at low levels by tumour cells themselves and/or by stromal cells in the mucosa. For instance, integrin signalling can promote the abundance of Wnt receptors at the plasma membrane.56 Alternatively, the availability of ligands of the Wnt pathway could be modulated by integrin signalling. This is less probable as we did not detect elevated Wnt ligand or sFRP expression in early tumour lesions with increased collagen I deposition (figure 3C, online supplemental figure 3B), although post-transcriptional regulation (eg, secretion) of these factors could be influenced by integrin signalling. We specifically highlight the role of Collagen I in regulating DGC cell fate, yet the contribution of other matrix components upregulated in DGC lesions requires further investigation.
Our findings support a model in which ECM remodelling sensitises cells to Wnt activation, sustaining the undifferentiated state of DGC cells within mucosal lesions. As tumour cells invade deeper gastric layers and move further from the stem cell niche, maintaining an undifferentiated state may in addition rely on cell-autonomous production of receptor ligands, potentiated by sFRPs. Future studies could clarify how these mechanisms interplay during DGC progression, which may help in understanding the trajectory from early lesions to metastatic DGC. For instance, although DGC mouse models suggest that undifferentiated cells with stem cell capacity initiate DGC formation,13 31 it remains unclear whether progression primarily involves the expansion of existing intramucosal PDCs or also relies on tumour cell plasticity through SRC dedifferentiation.32 In both scenarios, Wnt signalling regulation is critical for controlling these cellular states. Our model explains the persistent presence of SRCs throughout all DGC stages, including metastasis.57 Heterogeneous collagen deposition and Wnt3A/sFRP2 upregulation would result in a fraction of tumour cells lacking these signals and subsequently differentiating into SRCs. This contrasts with Wnt pathway mutations, which render tumour cells permanently niche-independent.
The identification of molecular changes driving the stepwise progression of DGC not only deepens our understanding of its evolution but also holds promise for improving tumour malignancy prediction. Thereby, these insights could be pivotal for guiding clinical decisions in HDGC patients, helping prioritise prophylactic gastrectomy for those at high risk of developing aggressive disease. Simultaneously, they support precise monitoring and timely intervention, enabling endoscopic surveillance strategies for low-risk patients. Additionally, our findings highlight the therapeutic potential of targeting Wnt signalling and its upstream regulatory mechanisms to halt the development of advanced sporadic and HDGC, including Porcupine inhibitors that are currently under clinical evaluation.58 This expands the therapeutic scope of Wnt-pathway inhibition beyond tumours harbouring genetic alterations in its core components.
Supplemental material
Supplemental material
Data availability statement
Data are available on reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information. Normalised and averaged data of the Nanostring DSP analysis has been added as a supplementary table. Raw counts of the Nanostring DSP analysis and whole exome sequencing data are available on request.
Ethics statements
Patient consent for publication
Ethics approval
The study was approved by the Radboudumc Research Ethics Committee (2020-6617).
Acknowledgments
We thank the Useq facility (Ies Nijman and Robin Geene, UMCU, Utrecht, The Netherlands) for support with Nanostring GeoMx DSP, Remco van Cruchten (Radboudumc, Nijmegen, The Netherlands) for support in data analysis of whole exome sequencing, Elisa Vink-Borger and Natasja Rutgers (Radboudumc, Nijmegen, The Netherlands) for DNA isolation, Claudia Janda for providing us with Frizzled inhibitor (F2.I), Ingrid Jordens (UMCU, Utrecht, The Netherlands) for critical reading of the manuscript, and members of our laboratory for the helpful discussions throughout the project.
References
Footnotes
X @drcolinwood, @nigeljamieson, @@Gloerichlab
Contributors LJSK, RSvdP and MG conceived the study; LJSK, JM, MM, MV, AK, RSvdP and MG designed experiments; LJSK, JM, MM and MV performed experiments and analysis, CW and NBJ supported analysis, LAAB, LLK, JMvD and TMB provided and curated patient tissue. LJSK, RSvdP and MG wrote the manuscript. All other authors provided input on the manuscript. MG is the guarantor of the study. ChatGPT was used to proofread written text and correct grammar and improve sentence structure.
Funding This work was supported by the Dutch Cancer Foundation (KWF-12345 and the Netherlands Organization for Scientific Research (NWO; ZonMW 2023 clinical fellow 09032212110047, 016.Vidi.189.166, NWO gravitational program CancerGenomiCs.nl 024.001.028 and the Science-XL research program The Active Matter Physics of Collective Metastasis 2019.022).
Competing interests None declared.
Patient and public involvement Patients and/or the public were involved in the design, or conduct, or reporting, or dissemination plans of this research. Refer to the Methods section for further details.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.